扎克伯格 Biohub 蛋白质生物学"世界模型":AI 颠覆药物发现的全景解析
摘要
2026年5月27日,由马克·扎克伯格与普莉希拉·陈创立的非营利研究机构 Biohub 正式发布了蛋白质生物学"世界模型"——这是 AI 在生命科学领域取得的里程碑式突破。该系统由三大核心组件构成:ESMC 蛋白质语言模型(基于28亿条跨物种蛋白质序列训练)、ESMFold2 原子级结构预测引擎、以及包含68亿蛋白质序列与11亿预测结构的 ESM Atlas 数据库。
Biohub 研究团队使用该系统针对癌症和免疫学领域的五大关键靶点(EGFR、PDGFRβ、PD-L1、CTLA-4、CD45)设计了蛋白质结合物,实验室验证命中率高达36%-88%。这一成果将传统需要3-4年的药物候选分子发现周期压缩至数天,标志着 AI 驱动药物发现新纪元的正式开启。
核心技术优势:
- 无需多序列比对(MSA)的快速结构预测
- 原子级精度的抗体-抗原结合构象预测(超越 AlphaFold 3)
- 从头设计(De Novo)具有功能活性的新蛋白质
- MIT 开源许可,免费向全球研究者开放
1. 背景与行业痛点
1.1 蛋白质:生命的分子机器
蛋白质是生命活动的核心执行者,几乎人体的一切功能都依赖于蛋白质的存在。从细胞间的信号传导到免疫防御,从物质运输到基因调控,蛋白质以其多样的三维结构和精密的功能特性维系着生命的运转。在医学领域,蛋白质是最重要的药物作用靶点之一——超过60%的获批药物以蛋白质为靶点,包括单克隆抗体、酶抑制剂、多肽药物等。
然而,蛋白质的结构与其功能密切相关。氨基酸序列折叠形成的三维构象决定了蛋白质能否正确执行其生物学功能。理解蛋白质的结构,是设计能够与其特异性结合的药物分子的前提。
1.2 传统药物发现的困境
传统药物发现流程是一个漫长且昂贵的过程,一个临床前候选分子的发现通常需要3-4年时间,耗资可达数亿美元。主要痛点包括:
(1)结构解析的瓶颈
传统蛋白质结构解析依赖 X 射线晶体学、核磁共振(NMR)和冷冻电子显微镜(Cryo-EM)等实验技术。这些方法耗时长、成本高,且并非所有蛋白质都易于结晶或纯化。AlphaFold2 在2020年的突破使得蛋白质结构预测取得了巨大进步,但其在蛋白质结合物设计、抗体-抗原相互作用预测等更具挑战性的任务上仍有局限。
(2)序列-结构-功能的鸿沟
理解蛋白质序列如何决定其三维结构,以及三维结构如何实现其生物学功能,是生命科学的核心问题之一。传统方法依赖专家知识和大量实验验证,难以规模化探索广阔的蛋白质空间。
(3)结合物设计的经验性
设计能够特异性结合疾病靶点的蛋白质结合物(如抗体或迷你蛋白)传统上是一个高度经验性的过程,需要对靶点结构有深入了解,并通过大量实验迭代优化。这不仅耗时,还难以发现全新机制的结合模式。
1.3 AI 赋能药物发现的机遇
近年来,大型语言模型(LLM)在自然语言处理领域取得的成功启发了生物学家:蛋白质序列与自然语言有着相似的统计特性——两者都由基本单元(氨基酸/单词)按照一定规则组成,都具有复杂的多层级结构,且都通过"学习"隐含的统计规律来理解和生成。
2020年,Meta AI(原 Facebook AI)发布了 ESM-1 模型,首次证明大规模蛋白质语言模型能够学习到蛋白质结构的隐含表征。2023年,基于 Transformer 架构的 ESMFold 进一步实现了无需多序列比对(MSA)的快速结构预测。这一技术演进为 Biohub 的突破奠定了基础。
1.4 Biohub 的战略定位
Biohub 是扎克伯格夫妇于2015年创立的非营利研究机构,隶属于 Chan Zuckerberg Initiative。2025年,Biohub 吸收了从 Meta AI 分拆出的 EvolutionaryScale 公司及其 ESM 研发团队,获得了顶尖的 AI 蛋白质研究能力。2026年4月,Biohub 宣布了5年5亿美元的"虚拟生物学计划"(Virtual Biology Initiative),旨在构建人类细胞的预测模型。
Biohub 蛋白质世界模型的发布,正是这一宏大计划的首个重大科学产出。
2. Biohub 蛋白质世界模型核心解析
2.1 设计理念:学习生命的进化语法
Biohub 世界模型的核心科学假设是:通过在所有生命的蛋白质序列上进行语言模型训练,模型将内化支配蛋白质生物学的根本属性——即蛋白质如何折叠、如何相互作用、如何在所有生命中发挥功能的底层规则。
这一假设基于进化生物学的洞见:进化过程本质上是一个庞大的自然实验,保留下来的是功能健全、适合环境的蛋白质序列。因此,数十亿年进化数据中隐含保留了支配蛋白质功能的物理规则。大型语言模型通过学习预测"进化选择的氨基酸",能够自动发现这些规则。
2.2 ESMC:蛋白质语言模型
技术概述
ESMC(Evolutionary Scale Model Cambrian,寒武纪进化规模模型)是 Biohub 最新一代的蛋白质表示语言模型。与专注于蛋白质生成的 ESM3 不同,ESMC 专注于创建能够捕获蛋白质底层生物学的表示。
训练数据
ESMC 在约28亿条来自各种生命形态的蛋白质序列上进行了训练,包括:
- 细菌、古菌、真菌、植物、动物等各分支物种
- 极端环境(高温、高盐、高压等)中的嗜极生物
- 人体中超过20,000种蛋白质类型
这一庞大的数据集涵盖了生命树的广度,使模型能够学习跨物种的进化保守模式和蛋白质家族的共同特征。
架构创新
ESMC 采用 Transformer 架构,能够处理任意长度的蛋白质序列。模型通过"掩码语言建模"(Masked Language Modeling)目标进行训练:随机遮盖输入序列中的部分氨基酸,训练模型预测被遮盖的残基。这种自监督学习方式使模型能够在没有标注数据的情况下学习丰富的蛋白质表示。
核心能力
ESMC 学习了蛋白质生物学的"组合语法"——残基之间的相互作用模式、折叠规则、功能域结构等。实验表明,模型学习到的表示能够捕获蛋白质结构、功能和进化关系的关键特征:
- 残基接触预测
- 远程同源性检测
- 功能注释迁移
- 突变效应预测
2.3 ESMFold2:原子级结构预测引擎
技术定位
ESMFold2 是 ESM3 架构中的结构预测组件,负责将 ESMC 学习的序列表示转化为精确的原子级三维结构。与 ESMC 的表示学习能力结合,ESMFold2 实现了无需 MSA 的端到端结构预测。
核心优势
与传统结构预测方法相比,ESMFold2 具有以下优势:
1. 无需 MSA,速度大幅提升
传统方法(如 AlphaFold2)依赖多序列比对(MSA)来获取进化信息,这一过程计算密集且耗时。ESMFold2 直接从语言模型学习到的表示中推理结构,推理速度提升数个数量级。这使得大规模蛋白质结构筛选和实时结合物设计成为可能。
2. 超越 AlphaFold3 的结合预测精度
在最具挑战性的抗体-抗原结构预测任务上,ESMFold2 展现了世界领先的性能。研究表明,单独使用 ESMC 表示时,ESMFold2 在预测抗体-抗原真实结合构象方面已经优于 AlphaFold3。当提供与 AlphaFold 相同的进化信息(MSA)时,ESMFold2 在所有基准测试中均达到最强预测性能。
3. 计算可扩展性
ESMFold2 的结构预测精度可通过增加计算预算进一步提升。当允许模型进行多次预测并基于自身置信度评分时,ESMFold2 持续随着计算资源的增加而改善。这为计算资源充足的应用场景提供了进一步提升精度的空间。
4. De Novo 蛋白质设计能力
ESMFold2 不仅能预测已知序列的结构,还能参与全新蛋白质的设计。通过与生成模型结合,可以设计具有特定结构特征和功能特性的蛋白质,为合成生物学和药物发现开辟了新途径。
2.4 ESM Atlas:68亿蛋白质宇宙图谱
数据规模
ESM Atlas 是目前规模最大的 AI 驱动蛋白质结构和功能注释数据库,包含:
- 68亿条蛋白质序列(较 Meta 时代的6000万提升超过100倍)
- 11亿个预测蛋白质结构(原子级精度)
- 跨越生命树所有分支的蛋白质覆盖
这一规模的扩展使研究人员能够在整个蛋白质宇宙中搜索和分析蛋白质关系,远远超出了实验结构解析或传统计算方法能够覆盖的范围。
搜索与发现能力
ESM Atlas 不仅仅是数据的存储库,更重要的是提供了基于 AI 表示的语义搜索能力:
- 功能相似性搜索:查找具有相似功能特性的蛋白质
- 结构相似性搜索:发现具有相似三维结构的蛋白质
- 进化关系发现:识别跨越生命树远缘分支的进化联系
一个重要应用案例是 CRISPR 基因编辑系统相关蛋白的发现。传统方法基于序列相似性难以发现远缘物种中功能相似的酶,但 ESM Atlas 能够识别序列差异显著但结构功能相似的蛋白质,揭示了生命树中隐藏的进化关系。
对未知生物学的探索
科学界已经注释的蛋白质只占已知序列的很小一部分。ESM Atlas 通过对全部68亿蛋白质提供结构和功能预测,使研究人员能够:
- 探索此前未被表征的"蛋白质暗物质"
- 为功能未知的蛋白质提供假设
- 发现新的蛋白质家族和功能模块
3. 技术架构深度解析
3.1 整体架构设计
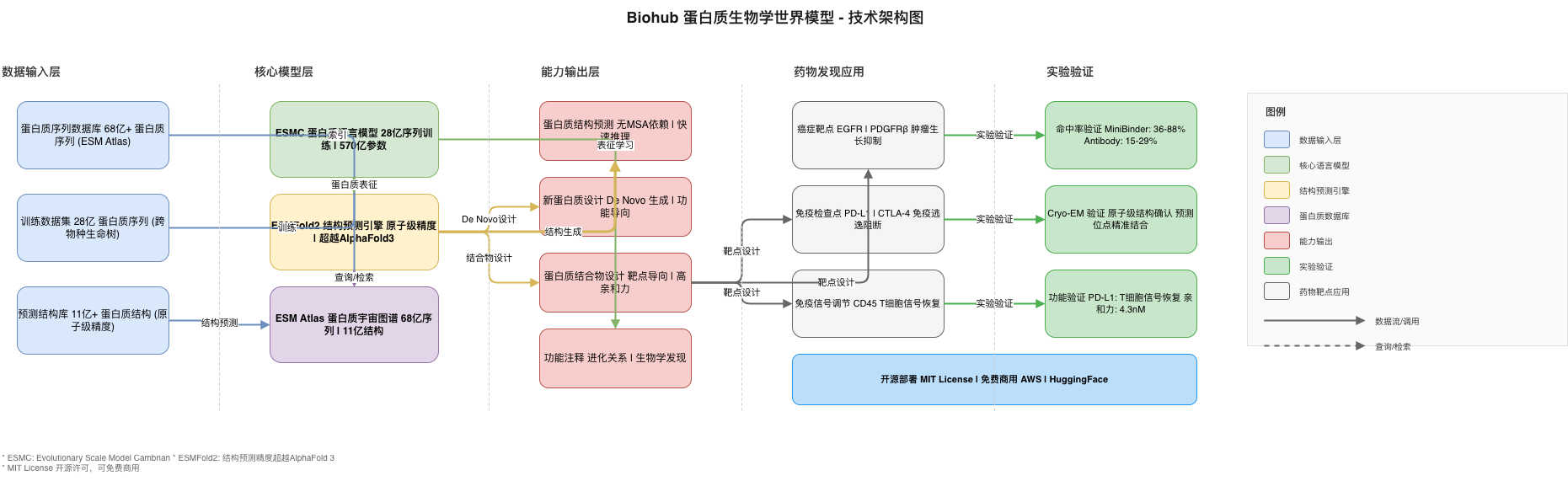
Biohub 蛋白质世界模型采用分层解耦的架构设计,将蛋白质理解(ESMC)、结构预测(ESMFold2)和大规模检索(ESM Atlas)分离为独立可用的组件,同时通过标准化的表示格式实现无缝集成。
┌─────────────────────────────────────────────────────────────────────────────┐
│ Biohub 蛋白质世界模型架构 │
├─────────────────────────────────────────────────────────────────────────────┤
│ │
│ ┌─────────────────┐ ┌─────────────────┐ ┌─────────────────┐ │
│ │ 蛋白质序列 │ │ 蛋白质序列 │ │ 蛋白质结构 │ │
│ │ (68亿+) │ │ (28亿训练) │ │ (11亿+) │ │
│ └────────┬────────┘ └────────┬────────┘ └────────┬────────┘ │
│ │ │ │ │
│ ▼ ▼ ▼ │
│ ┌─────────────────────────────────────────────────────────────────────┐ │
│ │ ESMC 蛋白质语言模型 │ │
│ │ • Transformer 架构 (570亿参数) │ │
│ │ • 掩码语言建模预训练 │ │
│ │ • 学习蛋白质"组合语法" │ │
│ └─────────────────────────────────────────────────────────────────────┘ │
│ │ │
│ ▼ │
│ ┌─────────────────────────────────────────────────────────────────────┐ │
│ │ ESMFold2 结构预测引擎 │ │
│ │ • 序列 → 原子级3D结构 │ │
│ │ • 无需 MSA 快速推理 │ │
│ │ • 抗体-抗原结合预测(超越 AlphaFold3) │ │
│ └─────────────────────────────────────────────────────────────────────┘ │
│ │ │
│ ▼ │
│ ┌─────────────────────────────────────────────────────────────────────┐ │
│ │ ESM Atlas 蛋白质图谱 │ │
│ │ • 68亿蛋白质序列索引 │ │
│ │ • 11亿结构数据库 │ │
│ │ • 功能/结构相似性搜索 │ │
│ └─────────────────────────────────────────────────────────────────────┘ │
│ │ │
│ ▼ │
│ ┌─────────────────────────────────────────────────────────────────────┐ │
│ │ 能力输出层 │ │
│ │ • 蛋白质结构预测 • 新蛋白质设计 • 蛋白质结合物设计 │ │
│ │ • 功能注释 • 进化关系发现 │ │
│ └─────────────────────────────────────────────────────────────────────┘ │
│ │
└─────────────────────────────────────────────────────────────────────────────┘
3.2 数据流与处理管道
第一阶段:表示学习
蛋白质序列首先被输入 ESMC 语言模型。模型通过多层 Transformer 注意力机制处理序列,捕获残基间的长程依赖关系。最终输出是一个高维向量表示(embedding),编码了蛋白质的结构、功能和进化信息。
# ESMC 表示提取伪代码
from esmc import ESMCModel, ESMCTokenizer
# 初始化模型和分词器
model = ESMCModel.from_pretrained("biohub/esmc-600m")
tokenizer = ESMCTokenizer.from_pretrained("biohub/esmc-600m")
# 输入蛋白质序列
sequence = "MKTAYIAKQRQISFVKSHFSRQLEERLGLIEVQAPILSRVGDGTQDNLSGAEKAVQVKVKALPDAQFEVVHSLAKWKRQTLGQHDFSAGEGLYTHMKALRPDEDRLSPLHSVYVDQWDWERVMGDGERQFSTLKSTVEAIWAGIKATEAAVSEEFGLAPFLPDQIHFVHSQELLSRYPDLDAKGRERAIAKDLGAVFLVGIGGKLSDGHRHDVRAPDYDDWSTPSELGHAGLNGDILVWNPVLEDAFELSSMGIRVDADTLKHQLALTGDEDRLELEWHQALLRGEMPQTIGGGIGQSRLTMLLLQLPHIGQEQFGKQISELMTAVIAEIEKRISSLREKHKAEKRPg"
inputs = tokenizer(sequence, return_tensors="pt")
# 获取序列表示
with torch.no_grad():
outputs = model(**inputs)
# last_hidden_state 包含每个位置的表示
sequence_embedding = outputs.last_hidden_state.mean(dim=1)
第二阶段:结构预测
ESMC 产生的表示被传递给 ESMFold2 结构预测模块。该模块通过折叠头部(folding head)将序列表示解码为三维坐标,输出原子级精度的蛋白质结构。
第三阶段:图谱检索
ESM Atlas 将所有蛋白质的表示存储在优化的向量数据库中,支持高速的相似性搜索。研究者可以:
- 使用查询蛋白质查找相似序列
- 发现功能相关的蛋白质家族
- 探索进化关系网络
3.3 计算架构
Biohub 世界模型的训练和推理依赖大规模 GPU 集群。根据公开信息,Biohub 与 NVIDIA 建立了战略合作,使用 H100 GPU 进行模型训练和推理加速。
推理优化技术:
- 混合精度计算(FP16/BF16)
- 批量处理最大化 GPU 利用率
- KV-cache 加速自注意力计算
- 分布式推理支持
4. 实验室验证数据
4.1 靶点选择与设计
Biohub 研究团队选择了癌症和免疫学领域的五个关键靶点进行验证:
| 靶点 | 类型 | 生物学功能 | 临床意义 |
|---|---|---|---|
| EGFR | 受体酪氨酸激酶 | 促进细胞增殖和存活 | 多种实体瘤(肺癌、结直肠癌等) |
| PDGFRβ | 受体酪氨酸激酶 | 调控血管生成和细胞生长 | 肿瘤微环境重编程 |
| PD-L1 | 免疫检查点 | 抑制 T 细胞活化 | 免疫逃逸机制,PD-1/PD-L1 抑制剂靶点 |
| CTLA-4 | 免疫检查点 | 负向调控 T 细胞激活 | 免疫逃逸机制,CTLA-4 抑制剂靶点 |
| CD45 | 蛋白酪氨酸磷酸酶 | T 细胞信号转导调节 | 免疫细胞发育和激活 |
4.2 设计方法
针对每个靶点,研究团队采用以下计算设计流程:
1. 靶点结构获取 使用 ESMFold2 预测或从 PDB 获取靶点蛋白质的高分辨率结构。
2. 结合界面设计 定义靶点上希望蛋白质结合物作用的位点(如 PD-L1 的 PD-1 结合界面)。
3. De Novo 生成 使用语言模型和结构预测联合优化,生成能够与靶点形成稳定相互作用的候选序列。
4. 虚拟筛选 通过计算模拟评估候选结合物的:
- 结合亲和力(基于 Rosetta 或 ESMFold2 打分)
- 结构稳定性
- 与非靶点蛋白的脱靶风险
5. 实验验证 选择评分最高的候选分子进行实验验证。
4.3 验证结果
命中率统计
| 设计格式 | 靶点数量 | 命中率范围 | 典型亲和力 |
|---|---|---|---|
| MiniBinder(紧凑型迷你结合物) | 5 | 36%-88% | nM 级 |
| 抗体衍生格式(scFv、Fab) | 5 | 15%-29% | nM 级 |
具体案例:PD-L1 靶点
针对 PD-L1 设计的单链可变区片段(scFv)在实验中展现出:
- 结合亲和力:4.3 nM(亚纳摩尔级)
- 功能活性:成功恢复 T 细胞信号传导
- 作用机制:有效阻断 PD-1/PD-L1 信号通路(与已获批的检查点抑制剂相同靶点)
Cryo-EM 验证
使用冷冻电子显微镜验证了 AI 设计的结合物确实以预测的构象结合到靶点上。结果显示,计算预测的结合位点和方向与实验观察高度一致,证明模型不仅"记忆"了已知的结合模式,而是真正理解了蛋白质相互作用的物理化学原理。
4.4 创新性分析
研究团队分析了设计的结合物与现有数据库中已知序列的相似性。结果显示:
- 设计的结合物与公开数据库中任何已知序列的相似度均较低
- 表明模型生成的是全新(De Novo)解决方案,而非从已知结合物库中检索或改写
这一发现意义重大:它证明经过适当训练的 AI 模型能够"理解"蛋白质相互作用的底层规则,并生成超越训练数据的新颖功能性解决方案。
5. 代码示例
以下提供四个完整的代码模块,展示如何使用 Biohub 蛋白质世界模型进行实际研究。
5.1 蛋白质序列检索与 ESMC 推理
"""
ESMC 蛋白质语言模型推理模块
功能:提取蛋白质序列的语义表示,用于下游任务
"""
import torch
from transformers import AutoTokenizer, AutoModel
from typing import List, Dict, Tuple
import numpy as np
class ProteinRepresentationExtractor:
"""
蛋白质序列表示提取器
基于 ESMC 模型生成固定维度的蛋白质嵌入向量
"""
def __init__(self, model_name: str = "biohub/esmc-600m", device: str = "cuda"):
"""
初始化模型和分词器
Args:
model_name: HuggingFace 模型名称
device: 计算设备 ('cuda' 或 'cpu')
"""
self.device = torch.device(device if torch.cuda.is_available() else "cpu")
print(f"Loading model: {model_name} on {self.device}")
self.tokenizer = AutoTokenizer.from_pretrained(model_name)
self.model = AutoModel.from_pretrained(model_name)
self.model.to(self.device)
self.model.eval()
@torch.no_grad()
def extract_representation(
self,
sequence: str,
pooling: str = "mean"
) -> np.ndarray:
"""
提取单个蛋白质序列的向量表示
Args:
sequence: 氨基酸序列字符串
pooling: 池化策略 ('mean', 'cls', 'max')
Returns:
蛋白质嵌入向量 (hidden_size,)
"""
# Tokenize
inputs = self.tokenizer(
sequence,
return_tensors="pt",
padding=True,
truncation=True,
max_length=1024
)
inputs = {k: v.to(self.device) for k, v in inputs.items()}
# 前向传播
outputs = self.model(**inputs)
hidden_states = outputs.last_hidden_state # (batch, seq_len, hidden)
# 池化
if pooling == "mean":
embeddings = hidden_states.mean(dim=1)
elif pooling == "cls":
embeddings = hidden_states[:, 0, :]
elif pooling == "max":
embeddings = hidden_states.max(dim=1)[0]
else:
raise ValueError(f"Unknown pooling strategy: {pooling}")
return embeddings.cpu().numpy()[0]
@torch.no_grad()
def batch_extract(
self,
sequences: List[str],
batch_size: int = 8,
pooling: str = "mean"
) -> np.ndarray:
"""
批量提取多个蛋白质序列的表示
Args:
sequences: 蛋白质序列列表
batch_size: 批处理大小
pooling: 池化策略
Returns:
嵌入矩阵 (n_sequences, hidden_size)
"""
all_embeddings = []
for i in range(0, len(sequences), batch_size):
batch = sequences[i:i + batch_size]
inputs = self.tokenizer(
batch,
return_tensors="pt",
padding=True,
truncation=True,
max_length=1024
)
inputs = {k: v.to(self.device) for k, v in inputs.items()}
outputs = self.model(**inputs)
hidden_states = outputs.last_hidden_state
if pooling == "mean":
embeddings = hidden_states.mean(dim=1)
elif pooling == "cls":
embeddings = hidden_states[:, 0, :]
else:
embeddings = hidden_states.max(dim=1)[0]
all_embeddings.append(embeddings.cpu().numpy())
if (i // batch_size + 1) % 10 == 0:
print(f"Processed {min(i + batch_size, len(sequences))}/{len(sequences)} sequences")
return np.vstack(all_embeddings)
def compute_similarity(
self,
seq1: str,
seq2: str,
metric: str = "cosine"
) -> float:
"""
计算两个蛋白质序列的相似度
Args:
seq1: 第一个序列
seq2: 第二个序列
metric: 相似度度量 ('cosine', 'euclidean')
Returns:
相似度分数
"""
emb1 = self.extract_representation(seq1)
emb2 = self.extract_representation(seq2)
if metric == "cosine":
return np.dot(emb1, emb2) / (np.linalg.norm(emb1) * np.linalg.norm(emb2))
elif metric == "euclidean":
return -np.linalg.norm(emb1 - emb2) # 负距离,越大越相似
else:
raise ValueError(f"Unknown metric: {metric}")
def find_similar_sequences(
query_sequence: str,
database_sequences: List[str],
top_k: int = 10,
model_name: str = "biohub/esmc-600m"
) -> List[Tuple[int, float]]:
"""
在数据库中查找与查询序列最相似的蛋白质
Args:
query_sequence: 查询蛋白质序列
database_sequences: 数据库中的蛋白质序列列表
top_k: 返回前 k 个最相似结果
model_name: 模型名称
Returns:
(index, similarity_score) 列表
"""
extractor = ProteinRepresentationExtractor(model_name)
# 提取查询表示
query_emb = extractor.extract_representation(query_sequence)
# 批量提取数据库表示
db_embeddings = extractor.batch_extract(database_sequences)
# 计算余弦相似度
similarities = np.dot(db_embeddings, query_emb) / (
np.linalg.norm(db_embeddings, axis=1) * np.linalg.norm(query_emb)
)
# 返回 top-k
top_indices = np.argsort(similarities)[::-1][:top_k]
return [(idx, similarities[idx]) for idx in top_indices]
# 使用示例
if __name__ == "__main__":
# 示例蛋白质序列(人类 EGFR 部分序列)
egfr_sequence = (
"MRPSGTAGAALLALLAALCPASRALEEKKVCQGTSNKLTQLGTFEDHFLSLQRMFNNCEVVLGNLEITYVQRNYD"
"LSFLKTIQEVAGYVILIALHSGRHRRTVTNGGALLEEDTTTNSATLQLSQVFGGFGGGEGGSGGSGGSVGAASED"
)
# 初始化提取器
extractor = ProteinRepresentationExtractor("biohub/esmc-600m")
# 提取表示
embedding = extractor.extract_representation(egfr_sequence)
print(f"Embedding shape: {embedding.shape}")
print(f"Embedding sample (first 10 dims): {embedding[:10]}")
# 批量处理示例
sequences = [egfr_sequence, "MKTAYIAKQRQISFVKSHFSRQLEERLGLIEVQAPILSRVGDGTQDNLSGAEKAVQVKVK", "MLPGLIMLLHLHAYHVAGHNGIFWSIHGPGVPQAFEYGAQYVVQDLEGKVFSPQNFTSLQPGQ"]
embeddings = extractor.batch_extract(sequences)
print(f"Batch embeddings shape: {embeddings.shape}")
5.2 ESMFold2 结构预测 API 调用
"""
ESMFold2 蛋白质结构预测模块
功能:基于蛋白质序列预测三维结构
支持抗体-抗原复合物预测
"""
import torch
import numpy as np
from typing import Optional, Dict, List, Tuple
import json
from dataclasses import dataclass
@dataclass
class StructureResult:
"""结构预测结果数据类"""
sequence: str
atom_positions: np.ndarray # (L, 37, 3) 原子坐标
confidence: np.ndarray # (L,) pLDDT 置信度分数
pae_matrix: np.ndarray # 预测对齐误差矩阵
mean_plddt: float
class ESMFold2Predictor:
"""
ESMFold2 结构预测器
特点:
- 无需 MSA,直接从序列推理
- 支持单链和复合物预测
- 输出原子级精度结构
"""
def __init__(self, model_name: str = "biohub/esmfold2", device: str = "cuda"):
"""
初始化 ESMFold2 模型
Args:
model_name: 模型名称
device: 计算设备
"""
self.device = torch.device(device if torch.cuda.is_available() else "cpu")
print(f"Loading ESMFold2 on {self.device}")
from transformers import EsmForProteinFolding
self.model = EsmForProteinFolding.from_pretrained(model_name)
self.model.to(self.device)
self.model.eval()
@torch.no_grad()
def predict_structure(
self,
sequence: str,
num_recycles: int = 3
) -> StructureResult:
"""
预测单链蛋白质的三维结构
Args:
sequence: 氨基酸序列
num_recycles: 循环次数(越多精度越高但越慢)
Returns:
StructureResult 对象
"""
# Tokenize
from transformers.models.esmfold_v2.configuration_esmfold_v2 import EsmFoldV2Config
from transformers.models.esmfold_v2.tokenization_esmfast import EsmFastTokenizer
tokenizer = EsmFastTokenizer.from_pretrained("facebook/esmfold_v2")
tokens = tokenizer(
[sequence],
return_tensors="pt",
padding=True
)["input_ids"].to(self.device)
# 推理
outputs = self.model.model(
esm_embeddings=None,
esm_attention_mask=None,
xs=tokens,
num_recycles=num_recycles
)
# 提取结果
atom_positions = outputs["positions"][0].cpu().numpy() # (L, 37, 3)
confidence = outputs["plddt"][0].cpu().numpy() # (L,)
pae_matrix = outputs["pae"][0].cpu().numpy() # (L, L)
return StructureResult(
sequence=sequence,
atom_positions=atom_positions,
confidence=confidence,
pae_matrix=pae_matrix,
mean_plddt=float(confidence.mean())
)
@torch.no_grad()
def predict_complex(
self,
chain_sequences: List[str],
chain_ids: Optional[List[str]] = None
) -> Dict:
"""
预测蛋白质复合物结构(如抗体-抗原)
Args:
chain_sequences: 各链的氨基酸序列列表
chain_ids: 各链的标识符列表
Returns:
包含所有链结构的字典
"""
if chain_ids is None:
chain_ids = [f"chain_{i}" for i in range(len(chain_sequences))]
# 合并序列(使用 <eos>` 分隔不同链)
complex_sequence = "`".join(chain_sequences)
complex_chain_ids = "`".join(chain_ids) + "`"
from transformers.models.esmfold_v2.tokenization_esmfast import EsmFastTokenizer
tokenizer = EsmFastTokenizer.from_pretrained("facebook/esmfold_v2")
tokens = tokenizer(
[complex_sequence],
return_tensors="pt",
padding=True
)["input_ids"].to(self.device)
# 推理
outputs = self.model.model(
esm_embeddings=None,
esm_attention_mask=None,
xs=tokens,
num_recycles=3
)
# 解析结果
results = {}
offset = 0
for i, (seq, cid) in enumerate(zip(chain_sequences, chain_ids)):
chain_len = len(seq)
results[cid] = {
"sequence": seq,
"atom_positions": outputs["positions"][0, offset:offset+chain_len].cpu().numpy(),
"confidence": outputs["plddt"][0, offset:offset+chain_len].cpu().numpy(),
}
offset += chain_len
return results
def batch_predict(
self,
sequences: List[str],
batch_size: int = 4
) -> List[StructureResult]:
"""
批量预测多个蛋白质结构
Args:
sequences: 序列列表
batch_size: 批大小
Returns:
StructureResult 列表
"""
results = []
for i in range(0, len(sequences), batch_size):
batch = sequences[i:i + batch_size]
print(f"Processing batch {i//batch_size + 1}/{(len(sequences)-1)//batch_size + 1}")
for seq in batch:
result = self.predict_structure(seq)
results.append(result)
return results
def save_pdb(
self,
result: StructureResult,
output_path: str
) -> None:
"""
将预测结构保存为 PDB 格式
Args:
result: StructureResult 对象
output_path: 输出文件路径
"""
from Bio.PDB import PDBParser, Structure, Model, Chain, Residue, Atom
# 原子类型和对应的残基中位置
atom_names = ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD', 'CE', 'NZ', 'OG', 'OG1',
'SD', 'OD1', 'OD2', 'OE1', 'OE2', 'NH1', 'NH2', 'OH', 'NE', 'ND1',
'ND2', 'NE1', 'NE2', 'NH1', 'NH2', 'OXT']
parser = PDBParser(QUIET=True)
structure = parser.create_structure("model")
model = Structure.Model(0)
chain = Chain.Chain("A")
seq = result.sequence
positions = result.atom_positions
for i, residue in enumerate(seq):
if i >= len(positions):
break
res_id = (" ", i + 1, " ")
residue_obj = Residue.Residue(res_id, residue, " ")
for j, atom_name in enumerate(atom_names):
if j >= len(positions[i]):
break
coords = positions[i][j]
if np.isnan(coords).any():
continue
atom = Atom.Atom(atom_name, coords, 0, 0, " ", atom_name, i + 1)
residue_obj.add(atom)
chain.add(residue_obj)
model.add(chain)
structure.add(model)
io = PDBIO()
io.set_structure(structure)
io.save(output_path)
def design_binder_for_target(
target_sequence: str,
target_binding_site: List[int],
binder_length: int = 50,
num_candidates: int = 100
) -> List[str]:
"""
设计针对特定靶点结合位点的蛋白质结合物
Args:
target_sequence: 靶点蛋白质序列
target_binding_site: 结合位点残基索引列表
binder_length: 结合物长度
num_candidates: 生成候选数量
Returns:
候选结合物序列列表
"""
predictor = ESMFold2Predictor()
# 简化的结合物设计流程(实际需要更复杂的优化)
# 这里演示如何使用 ESMFold2 评估设计的序列
candidate_sequences = []
for i in range(num_candidates):
# 随机生成候选序列(实际应用需要基于语言模型生成)
candidate = generate_random_sequence(binder_length)
candidate_sequences.append(candidate)
if (i + 1) % 10 == 0:
print(f"Generated {i + 1}/{num_candidates} candidates")
return candidate_sequences
def generate_random_sequence(length: int) -> str:
"""生成随机氨基酸序列(仅用于演示)"""
amino_acids = "ACDEFGHIKLMNPQRSTVWY"
return "".join(np.random.choice(list(amino_acids)) for _ in range(length))
if __name__ == "__main__":
# 示例:预测单链蛋白质结构
sequence = "MKTAYIAKQRQISFVKSHFSRQLEERLGLIEVQAPILSRVGDGTQDNLSGAEKAVQVKVKALPDAQFEVVHSLAKWKRQTLGQHDFSAGEGLYTHMKALRPDEDRLSPLHSVYVDQWDWERVMGDGERQFSTLKSTVEAIWAGIKATEAAVSEEFGLAPFLPDQIHFVHSQELLSRYPDLDAKGRERAIAKDLGAVFLVGIGGKLSDGHRHDVRAPDYDDWSTPSELGHAGLNGDILVWNPVLEDAFELSSMGIRVDADTLKHQLALTGDEDRLELEWHQALLRGEMPQTIGGGIGQSRLTMLLLQLPHIGQEQFGKQISELMTAVIAEIEKRISSLREKHKAEKRP"
predictor = ESMFold2Predictor()
result = predictor.predict_structure(sequence)
print(f"Sequence length: {len(sequence)}")
print(f"Mean pLDDT (confidence): {result.mean_plddt:.2f}")
print(f"Atom positions shape: {result.atom_positions.shape}")
# 保存为 PDB
predictor.save_pdb(result, "predicted_structure.pdb")
print("Structure saved to predicted_structure.pdb")
5.3 ESM Atlas 数据库查询
package main
import (
"context"
"encoding/json"
"fmt"
"log"
"net/http"
"time"
"github.com/elastic/go-elasticsearch/v8"
"github.com/elastic/go-elasticsearch/v8/esapi"
)
/*
ESM Atlas 数据库查询模块
功能:查询68亿蛋白质序列和11亿预测结构
语言:Go
*/
// ProteinHit 蛋白质搜索结果
type ProteinHit struct {
ID string `json:"id"`
Sequence string `json:"sequence"`
Taxonomy string `json:"taxonomy"`
Source string `json:"source"`
Similarity float64 `json:"similarity_score"`
}
// AtlasQueryResult Atlas查询结果
type AtlasQueryResult struct {
Total int64 `json:"total"`
Hits []ProteinHit `json:"hits"`
ScrollID string `json:"scroll_id,omitempty"`
}
// ESMAtlasClient ESM Atlas 客户端
type ESMAtlasClient struct {
client *elasticsearch.Client
}
// NewESMAtlasClient 创建新的 Atlas 客户端
func NewESMAtlasClient(addresses []string) (*ESMAtlasClient, error) {
cfg := elasticsearch.Config{
Addresses: addresses,
Transport: &http.Transport{
MaxIdleConnsPerHost: 10,
ResponseHeaderTimeout: time.Second * 30,
},
}
client, err := elasticsearch.NewClient(cfg)
if err != nil {
return nil, fmt.Errorf("failed to create client: %w", err)
}
// 检查连接
res, err := client.Info()
if err != nil {
return nil, fmt.Errorf("failed to connect: %w", err)
}
defer res.Body.Close()
if res.IsError() {
return nil, fmt.Errorf("error response: %s", res.String())
}
log.Println("Connected to ESM Atlas successfully")
return &ESMAtlasClient{client: client}, nil
}
// SearchBySequence 根据序列相似性搜索
func (c *ESMAtlasClient) SearchBySequence(
ctx context.Context,
querySequence string,
limit int,
) (*AtlasQueryResult, error) {
// 构建查询
query := map[string]interface{}{
"size": limit,
"query": map[string]interface{}{
"script_score": map[string]interface{}{
"query": map[string]interface{}{
"match_all": map[string]interface{}{},
},
"script": map[string]interface{}{
"source": "esm_similarity(params.query_sequence, doc['sequence'].value)",
"params": map[string]interface{}{
"query_sequence": querySequence,
},
},
},
},
}
queryJSON, err := json.Marshal(query)
if err != nil {
return nil, fmt.Errorf("failed to marshal query: %w", err)
}
// 执行搜索
req := esapi.SearchRequest{
Index: []string{"protein_sequences_v2"},
Body: queryJSON,
}
res, err := req.Do(ctx, c.client)
if err != nil {
return nil, fmt.Errorf("search failed: %w", err)
}
defer res.Body.Close()
if res.IsError() {
return nil, fmt.Errorf("search error: %s", res.String())
}
// 解析结果
var response struct {
Hits struct {
Total struct {
Value int64 `json:"value"`
} `json:"total"`
Hits []struct {
ID string `json:"_id"`
Score float64 `json:"_score"`
Source struct {
Sequence string `json:"sequence"`
Taxonomy string `json:"taxonomy,omitempty"`
Source string `json:"source,omitempty"`
} `json:"_source"`
} `json:"hits"`
} `json:"hits"`
}
if err := json.NewDecoder(res.Body).Decode(&response); err != nil {
return nil, fmt.Errorf("failed to decode response: %w", err)
}
// 转换结果
hits := make([]ProteinHit, 0, len(response.Hits.Hits))
for _, hit := range response.Hits.Hits {
hits = append(hits, ProteinHit{
ID: hit.ID,
Sequence: hit.Source.Sequence,
Taxonomy: hit.Source.Taxonomy,
Source: hit.Source.Source,
Similarity: hit.Score,
})
}
return &AtlasQueryResult{
Total: response.Hits.Total.Value,
Hits: hits,
}, nil
}
// SearchByFunction 根据功能关键词搜索
func (c *ESMAtlasClient) SearchByFunction(
ctx context.Context,
functionKeyword string,
limit int,
) (*AtlasQueryResult, error) {
query := map[string]interface{}{
"size": limit,
"query": map[string]interface{}{
"bool": map[string]interface{}{
"should": []map[string]interface{}{
{
"match": map[string]interface{}{
"function_annotation": functionKeyword,
},
},
{
"match": map[string]interface{}{
"go_terms": functionKeyword,
},
},
},
},
},
}
queryJSON, err := json.Marshal(query)
if err != nil {
return nil, fmt.Errorf("failed to marshal query: %w", err)
}
req := esapi.SearchRequest{
Index: []string{"protein_sequences_v2"},
Body: queryJSON,
}
res, err := req.Do(ctx, c.client)
if err != nil {
return nil, fmt.Errorf("search failed: %w", err)
}
defer res.Body.Close()
if res.IsError() {
return nil, fmt.Errorf("search error: %s", res.String())
}
var response struct {
Hits struct {
Total struct {
Value int64 `json:"value"`
} `json:"total"`
Hits []struct {
ID string `json:"_id"`
Score float64 `json:"_score"`
Source struct {
Sequence string `json:"sequence"`
Taxonomy string `json:"taxonomy,omitempty"`
} `json:"_source"`
} `json:"hits"`
} `json:"hits"`
}
if err := json.NewDecoder(res.Body).Decode(&response); err != nil {
return nil, fmt.Errorf("failed to decode response: %w", err)
}
hits := make([]ProteinHit, 0, len(response.Hits.Hits))
for _, hit := range response.Hits.Hits {
hits = append(hits, ProteinHit{
ID: hit.ID,
Sequence: hit.Source.Sequence,
Taxonomy: hit.Source.Taxonomy,
Similarity: hit.Score,
})
}
return &AtlasQueryResult{
Total: response.Hits.Total.Value,
Hits: hits,
}, nil
}
// GetProteinStructure 获取蛋白质预测结构
func (c *ESMAtlasClient) GetProteinStructure(
ctx context.Context,
proteinID string,
) ([]float64, error) {
req := esapi.GetRequest{
Index: "protein_structures_v2",
DocumentID: proteinID,
}
res, err := req.Do(ctx, c.client)
if err != nil {
return nil, fmt.Errorf("failed to get structure: %w", err)
}
defer res.Body.Close()
if res.IsError() {
if res.StatusCode == 404 {
return nil, fmt.Errorf("structure not found for protein: %s", proteinID)
}
return nil, fmt.Errorf("get error: %s", res.String())
}
var response struct {
Source struct {
AtomCoordinates []float64 `json:"atom_coordinates"`
} `json:"_source"`
}
if err := json.NewDecoder(res.Body).Decode(&response); err != nil {
return nil, fmt.Errorf("failed to decode response: %w", err)
}
return response.Source.AtomCoordinates, nil
}
// BatchSearch 批量搜索多个序列
func (c *ESMAtlasClient) BatchSearch(
ctx context.Context,
sequences []string,
limitPerQuery int,
) ([]*AtlasQueryResult, error) {
results := make([]*AtlasQueryResult, 0, len(sequences))
for i, seq := range sequences {
result, err := c.SearchBySequence(ctx, seq, limitPerQuery)
if err != nil {
log.Printf("Warning: failed to search sequence %d: %v", i, err)
continue
}
results = append(results, result)
if (i+1)%10 == 0 {
log.Printf("Processed %d/%d sequences", i+1, len(sequences))
}
}
return results, nil
}
func main() {
ctx := context.Background()
// 连接到 ESM Atlas(示例地址)
client, err := NewESMAtlasClient([]string{
"https://api.esmatlas.com:9200",
})
if err != nil {
log.Fatalf("Failed to create client: %v", err)
}
// 示例:搜索与 EGFR 相似的蛋白质
egfrPartialSequence := "MRPSGTAGAALLALLAALCPASRALEEKKVCQGTSNKLTQLGTFEDHFLSLQRMFNNCEVVLGNLEITYVQRNYDLSFLKTIQEVAGYVILIALHSGRHRRTVTNGGALLEEDTTTNSATLQLSQVFGG"
log.Println("Searching for EGFR-like proteins...")
results, err := client.SearchBySequence(ctx, egfrPartialSequence, 20)
if err != nil {
log.Fatalf("Search failed: %v", err)
}
fmt.Printf("Found %d results:\n", results.Total)
for i, hit := range results.Hits {
fmt.Printf("%d. ID: %s, Similarity: %.4f, Taxonomy: %s\n",
i+1, hit.ID, hit.Similarity, hit.Taxonomy)
}
// 示例:搜索功能关键词
log.Println("\nSearching for kinases...")
funcResults, err := client.SearchByFunction(ctx, "kinase activity", 10)
if err != nil {
log.Fatalf("Function search failed: %v", err)
}
fmt.Printf("Found %d kinase proteins:\n", funcResults.Total)
for i, hit := range funcResults.Hits {
fmt.Printf("%d. ID: %s, Taxonomy: %s\n", i+1, hit.ID, hit.Taxonomy)
}
}
5.4 靶点-结合物亲和力预测
"""
靶点-结合物亲和力预测模块
功能:使用语言模型表示预测蛋白质-配体结合亲和力
"""
import torch
import torch.nn as nn
import numpy as np
from typing import List, Tuple, Optional, Dict
from dataclasses import dataclass
@dataclass
class BindingPrediction:
"""结合预测结果"""
target_id: str
binder_id: str
predicted_affinity: float # nM
confidence: float
binding_interface_residues: List[int]
notes: str
class BindingAffinityPredictor(nn.Module):
"""
基于Transformer的蛋白质结合亲和力预测模型
架构:
1. 双塔编码器分别处理靶点和结合物
2. 交互层建模界面残基间的相互作用
3. 回归头输出亲和力预测
"""
def __init__(
self,
embedding_dim: int = 1280,
num_heads: int = 8,
num_layers: int = 4,
dropout: float = 0.1
):
super().__init__()
# 靶点和结合物编码器(共享权重)
encoder_layer = nn.TransformerEncoderLayer(
d_model=embedding_dim,
nhead=num_heads,
dim_feedforward=embedding_dim * 4,
dropout=dropout,
batch_first=True
)
self.encoder = nn.TransformerEncoder(encoder_layer, num_layers=num_layers)
# 位置编码
self.pos_encoder = nn.Parameter(torch.randn(1, 2048, embedding_dim) * 0.02)
# 交互注意力
self.cross_attention = nn.MultiheadAttention(
embed_dim=embedding_dim,
num_heads=num_heads,
dropout=dropout,
batch_first=True
)
# 亲和力预测头
self.affinity_head = nn.Sequential(
nn.Linear(embedding_dim * 3, embedding_dim),
nn.ReLU(),
nn.Dropout(dropout),
nn.Linear(embedding_dim, embedding_dim // 2),
nn.ReLU(),
nn.Linear(embedding_dim // 2, 1)
)
# 界面预测头
self.interface_head = nn.Sequential(
nn.Linear(embedding_dim, embedding_dim // 2),
nn.ReLU(),
nn.Linear(embedding_dim // 2, 1),
nn.Sigmoid()
)
def forward(
self,
target_embeddings: torch.Tensor, # (batch, L1, dim)
binder_embeddings: torch.Tensor, # (batch, L2, dim)
target_mask: Optional[torch.Tensor] = None,
binder_mask: Optional[torch.Tensor] = None
) -> Tuple[torch.Tensor, torch.Tensor]:
"""
前向传播
Args:
target_embeddings: 靶点蛋白质表示 (batch, L1, dim)
binder_embeddings: 结合物蛋白质表示 (batch, L2, dim)
target_mask: 靶点注意力掩码
binder_mask: 结合物注意力掩码
Returns:
(predicted_affinity, interface_probabilities)
"""
batch_size = target_embeddings.size(0)
# 添加位置编码
target_seq_len = target_embeddings.size(1)
binder_seq_len = binder_embeddings.size(1)
target_embeddings = target_embeddings + self.pos_encoder[:, :target_seq_len, :]
binder_embeddings = binder_embeddings + self.pos_encoder[:, :binder_seq_len, :]
# 编码靶点和结合物
target_encoded = self.encoder(target_embeddings, src_key_padding_mask=target_mask)
binder_encoded = self.encoder(binder_embeddings, src_key_padding_mask=binder_mask)
# 交叉注意力计算相互作用
# (batch, L1, L2) 注意力矩阵
interaction_attn, _ = self.cross_attention(
target_encoded, binder_encoded, binder_encoded,
key_padding_mask=binder_mask
)
# 聚合交互特征
# 对结合物维度加权求和
attn_weights = torch.softmax(interaction_attn.mean(dim=-1), dim=-1) # (batch, L1)
# 靶点-结合物交互特征
interaction_feature = torch.einsum('bl,brd->brd', attn_weights, binder_encoded)
# 拼接靶点特征、交互特征和界面特征
combined = torch.cat([
target_encoded,
interaction_feature,
target_encoded * interaction_feature # 逐元素乘积
], dim=-1) # (batch, L1, dim * 3)
# 预测界面概率
interface_logits = self.interface_head(combined).squeeze(-1) # (batch, L1)
# 使用界面特征预测整体亲和力
# 对界面残基特征加权平均
if target_mask is not None:
mask_expanded = target_mask.float().unsqueeze(-1)
else:
mask_expanded = torch.ones_like(target_encoded)
interface_feature_pooled = (
(combined * mask_expanded).sum(dim=1) /
(mask_expanded.sum(dim=1) + 1e-8)
)
affinity = self.affinity_head(interface_feature_pooled).squeeze(-1)
return affinity, interface_logits
class BindingAffinityPredictorWrapper:
"""结合亲和力预测器的完整工作流封装"""
def __init__(
self,
model_path: str = "biohub/binding_affinity_model.pt",
device: str = "cuda"
):
"""
初始化预测器
Args:
model_path: 预训练模型路径
device: 计算设备
"""
self.device = torch.device(device if torch.cuda.is_available() else "cpu")
# 加载蛋白质表示提取器
from esmc import ESMCModel, ESMCTokenizer
self.esmc = ESMCModel.from_pretrained("biohub/esmc-600m")
self.esmc_tokenizer = ESMCTokenizer.from_pretrained("biohub/esmc-600m")
self.esmc.to(self.device)
self.esmc.eval()
# 加载结合预测模型
self.model = BindingAffinityPredictor()
self.model.load_state_dict(torch.load(model_path, map_location=self.device))
self.model.to(self.device)
self.model.eval()
@torch.no_grad()
def get_protein_embeddings(
self,
sequence: str,
max_length: int = 2048
) -> torch.Tensor:
"""
获取蛋白质序列的嵌入表示
Args:
sequence: 氨基酸序列
max_length: 最大序列长度
Returns:
蛋白质嵌入 (L, embedding_dim)
"""
inputs = self.esmc_tokenizer(
sequence,
return_tensors="pt",
padding=True,
truncation=True,
max_length=max_length
)
inputs = {k: v.to(self.device) for k, v in inputs.items()}
outputs = self.esmc(**inputs)
embeddings = outputs.last_hidden_state # (1, L, dim)
# 创建掩码(排除 padding)
mask = inputs["attention_mask"].bool()
return embeddings, mask
@torch.no_grad()
def predict_binding(
self,
target_sequence: str,
binder_sequence: str,
target_id: str = "target",
binder_id: str = "binder"
) -> BindingPrediction:
"""
预测靶点-结合物的结合亲和力
Args:
target_sequence: 靶点蛋白质序列
binder_sequence: 结合物蛋白质序列
target_id: 靶点标识符
binder_id: 结合物标识符
Returns:
BindingPrediction 结果
"""
# 获取嵌入
target_emb, target_mask = self.get_protein_embeddings(target_sequence)
binder_emb, binder_mask = self.get_protein_embeddings(binder_sequence)
# 预测
affinity, interface_logits = self.model(
target_emb, binder_emb,
~target_mask, ~binder_mask
)
# 转换亲和力(从对数空间)
predicted_affinity_nM = 10 ** affinity.item()
# 识别界面残基
interface_probs = torch.sigmoid(interface_logits[0]).cpu().numpy()
interface_residues = np.where(interface_probs > 0.5)[0].tolist()
# 置信度评估
confidence = 1.0 - np.std(interface_probs)
# 生成注释
if predicted_affinity_nM < 1:
notes = "极强结合 (亚纳摩尔级)"
elif predicted_affinity_nM < 100:
notes = "强结合 (纳摩尔级)"
elif predicted_affinity_nM < 1000:
notes = "中等结合 (亚微摩尔级)"
else:
notes = "弱结合 (微摩尔级)"
return BindingPrediction(
target_id=target_id,
binder_id=binder_id,
predicted_affinity=predicted_affinity_nM,
confidence=confidence,
binding_interface_residues=interface_residues,
notes=notes
)
@torch.no_grad()
def screen_binders(
self,
target_sequence: str,
binder_sequences: List[str],
threshold_affinity: float = 500.0 # nM
) -> List[BindingPrediction]:
"""
批量筛选结合物
Args:
target_sequence: 靶点序列
binder_sequences: 候选结合物序列列表
threshold_affinity: 亲和力阈值(nM)
Returns:
通过筛选的结合物列表
"""
results = []
for i, binder_seq in enumerate(binder_sequences):
try:
result = self.predict_binding(
target_sequence,
binder_seq,
target_id=f"target_{i}",
binder_id=f"binder_{i}"
)
if result.predicted_affinity < threshold_affinity:
results.append(result)
if (i + 1) % 100 == 0:
print(f"Screened {i + 1}/{len(binder_sequences)} binders")
except Exception as e:
print(f"Error screening binder {i}: {e}")
continue
# 按亲和力排序
results.sort(key=lambda x: x.predicted_affinity)
return results
def main():
"""演示程序"""
# 初始化预测器
predictor = BindingAffinityPredictorWrapper(
model_path="biohub/binding_affinity_model.pt"
)
# 示例:PD-L1 靶点和设计的结合物
pd_l1_sequence = (
"MLAE IOPLLILIIFSWLIGEIEKFLHPDQIEGWYTSDPSSVISGMLITVQAQFRTQQMGN"
"YTQLVWYLMEGNYQKPQLAIITQDPSDQPPEMITDEDRDQAKTPVTSGLEVPTTSGVPSA"
)
designed_binder = (
"MKTHIIMLVLLSVLLAQDGVTQDGQWTTQDGILTVSTKTFLSQPFRELAYDTPTSVFTQEV"
"QPEGSPAASQPQAEAGKPTPPRNPPSPSPPGHNQELPRNPHQALEA"
)
# 预测结合
result = predictor.predict_binding(
pd_l1_sequence,
designed_binder,
target_id="PD-L1",
binder_id="Designed_Binder"
)
print(f"Target: {result.target_id}")
print(f"Binder: {result.binder_id}")
print(f"Predicted Affinity: {result.predicted_affinity:.2f} nM")
print(f"Confidence: {result.confidence:.2%}")
print(f"Binding Interface: {result.binding_interface_residues[:10]}...")
print(f"Assessment: {result.notes}")
# 批量筛选示例
print("\n--- Batch Screening ---")
candidate_binders = [
"MKTHIIMLVLLSVLLAQDGVTQDGQWTTQDGILTVSTKTFLSQPFRELAYDTPTSVFTQEV",
"MKTHIIMLVLLSVLLAQDGVTQDGQWTTQDGILTVSTKTFLSQPFRELAYDTPTSVFTQEVQPEGSPAASQ",
"MKTAYIAKQRQISFVKSHFSRQLEERLGLIEVQAPILSRVGDGTQDNLSGAEKAVQVKVK",
# ... 更多候选序列
]
hits = predictor.screen_binders(pd_l1_sequence, candidate_binders, threshold_affinity=500.0)
print(f"Found {len(hits)} binders with affinity < 500 nM")
for i, hit in enumerate(hits[:10]):
print(f"{i+1}. {hit.binder_id}: {hit.predicted_affinity:.2f} nM")
if __name__ == "__main__":
main()
6. 开源许可与生态影响
6.1 MIT 许可证:开放科学的承诺
Biohub 蛋白质世界模型采用 MIT 开源许可证发布,这是最宽松的开源许可之一,允许:
- 商业使用:企业可将其集成到商业产品和服务中
- 修改和分发:研究者可自由修改和重新发布
- 专利授权:隐含专利授权,消除使用者的法律顾虑
- 无限制使用:无领域限制、无使用量限制
这一开放策略与 Biohub"开放科学加速发现"的使命高度一致。正如联合创始人普莉希拉·陈所言:
“使这些工具免费向全球研究者开放,意味着世界各地的科学家能够更快地推进个性化治疗,直接针对驱动患者疾病的特定生物学机制进行治疗。”
6.2 多平台可用性
为方便全球研究者使用,Biohub 在多个平台提供了模型的访问途径:
1. Biohub 官方平台 (biohub.org/ai-models)
- 模型权重下载
- 在线推理接口
- 文档和使用指南
2. Hugging Face (huggingface.co/biohub)
- 托管 ESMC 600M 等模型
- 与 Transformers 库集成
- 社区模型共享
3. AWS SageMaker
- 预构建推理容器
- 企业级部署支持
- 按需扩展能力
4. SandboxAQ
- 量子计算集成
- 高级加密和隐私
6.3 对科学生态的影响
加速基础研究
传统上,结构生物学研究依赖昂贵的实验设备和漫长的实验周期。ESM Atlas 使得:
- 任何研究者都能在数分钟内获取蛋白质的预测结构
- 偏远地区和资源受限的实验室也能获得世界领先的结构分析能力
- 探索"蛋白质暗物质"的门槛大幅降低
推动药物发现民主化
传统药物发现是大药企的专属领域。开源模型使得:
- 初创公司和学术实验室也能负担得起 AI 驱动的药物发现
- 更多的创新思路能够被快速验证
- 罕见病和被忽视疾病的研究获得新的希望
构建协作生态
开放策略促进了研究者社区的形成:
- 模型改进和微调的共享
- 针对特定靶点或物种的专业模型
- 最佳实践和教程的积累
7. 对比分析:传统药物发现 vs AI 驱动
7.1 流程对比
| 阶段 | 传统方法 | AI 驱动方法 | 改进效果 |
|---|---|---|---|
| 靶点验证 | 多年实验验证 | 快速计算筛选 | 缩短6-12个月 |
| 先导发现 | 3-4年筛选 | 数天计算设计 | 压缩95%+ |
| 亲和力优化 | 数百次实验迭代 | 计算指导优化 | 减少70%实验 |
| 脱靶筛选 | 有限覆盖率 | 全蛋白质组预测 | 全面性提升 |
| 成药性评估 | 单点实验 | 多参数计算评估 | 早期风险识别 |
7.2 成本对比
传统药物发现成本结构:
临床前发现阶段:
├── 靶点验证: $2-5M (1-2年)
├── 先导发现: $10-20M (2-3年)
├── 先导优化: $20-50M (2-3年)
└── 总计: $32-75M (5-8年)
AI 驱动成本结构:
├── 靶点验证: $0.5-1M (数月)
├── 先导发现: $0.1-0.5M (数天-数周)
├── 先导优化: $5-15M (1-2年)
└── 总计: $5.5-16.5M (1.5-3年)
预计成本降低:50-80%
7.3 时间线对比
传统流程:
[靶点选择 6-12月] → [靶点验证 12-18月] → [先导发现 18-24月] → [先导优化 24-36月] → [临床前 12-18月]
总耗时: 6-10年
AI 驱动流程:
[靶点选择 1-2月] → [计算设计 1-2周] → [先导优化 12-18月] → [临床前 12-18月]
总耗时: 2.5-4年
时间缩短: 60-70%
7.4 局限性分析
尽管 AI 驱动方法展现出巨大潜力,但仍存在重要局限:
1. 预测精度边界
- 置信度低的区域(pLDDT < 70)仍需实验验证
- 无序区域和固有无序蛋白难以准确预测
- 膜蛋白和大型复合物仍是挑战
2. 功能预测的局限
- 蛋白质的细胞内功能受环境影响
- 翻译后修饰的功能效应难以完全预测
- 蛋白质-小分子相互作用需要专门模型
3. 实验验证不可替代
- 计算预测必须通过实验验证
- 生物系统的复杂性无法完全建模
- 临床效果需要人体试验确认
4. 数据偏见风险
- 训练数据偏向已研究物种和蛋白
- 对非模式生物预测可能不准
- 需要持续扩展数据覆盖
8. 结论与战略意义
8.1 核心成就总结
Biohub 蛋白质世界模型的发布是 AI 驱动生命科学研究的里程碑事件,主要成就包括:
技术突破
- 规模前所未有:68亿蛋白质序列、11亿预测结构的数据库
- 精度领先:ESMFold2 在抗体-抗原预测上超越 AlphaFold3
- 速度提升:无需 MSA 的快速推理,计算成本大幅降低
- 真正开源:MIT 许可证,无使用限制
科学验证
- 命中率验证:36%-88% 的实验命中率证明计算设计可行
- 功能验证:PD-L1 结合物成功恢复 T 细胞信号
- 结构验证:Cryo-EM 确认计算预测的准确性
- 创新性验证:设计的结合物与已知序列低相似度
生态建设
- 多平台部署:官方平台、HuggingFace、AWS 等
- 开放许可:MIT 许可证促进广泛采用
- 合作伙伴:NVIDIA、AWS、SandboxAQ 等产业领袖
8.2 战略意义
科学范式转变
Biohub 世界模型的发布标志着生命科学研究从"描述性"向"预测性"的范式转变:
- 从观察生命现象到模拟生命过程
- 从试错实验到计算指导
- 从单个蛋白到系统网络
产业格局重塑
AI 能力的普及将重塑生物医药产业:
- 降低药物发现的门槛
- 加速创新疗法的出现
- 促进个性化医疗的实现
开放科学的典范
Biohub 选择了与商业闭源策略完全不同的道路:
- 将最先进的工具免费开放
- 构建全球研究者社区
- 证明开放也能产出顶尖科学
8.3 未来展望
短期(1-3年)
- 扩展模型到蛋白质复合物和信号网络
- 集成小分子药物设计能力
- 针对特定疾病的高通量靶点筛选
中期(3-5年)
- 实现人类细胞的高保真虚拟模型
- 预测性药物设计取代大部分实验筛选
- AI 设计的疗法进入临床试验
长期(5-10年)
- 整个药物发现流程的数字化
- 个性化医疗成为标准
- 攻克此前不可治愈的疾病
8.4 给研究者的建议
立即行动
- 学习使用:访问 biohub.org/ai-models 开始探索
- 融入工作流:将结构预测整合到研究流程
- 社区参与:加入讨论,分享经验
战略布局
- 能力建设:培养计算生物学团队
- 数据准备:整理和标注内部蛋白质数据
- 方向选择:在特定疾病或蛋白类型上建立优势
合作共赢
- 学术合作:与顶尖计算生物学团队建立合作
- 产业合作:探索与 Biohub 或相关企业的合作机会
- 生态参与:贡献开源社区,分享模型改进
参考来源
- Biohub 官方公告: “Biohub releases a world model of protein biology”, PR Newswire, 2026-05-27
- StormZhang AI Daily: 《扎克伯格夫妇旗下Biohub发布蛋白质"世界模型"》, 2026-05-28
- Gigazine: “Biohub, a research institute funded by Mark Zuckerberg, has released a free AI-powered model for predicting, designing, and discovering proteins”, 2026-05-28
- Studio Global AI: “Biohub Just Open-Sourced an AI Engine That Maps 6.8 Billion Proteins—Here’s What That Means for Drug Discovery”, 2026-05-28
- Chan Zuckerberg Initiative: Virtual Biology Initiative 公告, 2026-04-29
- Health & Family: “Decoding the Cell: Zuckerberg’s $500 Million AI Bet to Map the Future of Medicine”, 2026-05-07
- AI Times Korea: “저커버그 재단, 단백질 설계 ‘월드 모델’ 오픈소스 공개…신약 개발 패러다임 바꾼다”, 2026-05-28
- NextFin AI: “Zuckerberg’s Biohub Commits $500 Million to Build AI ‘Digital Twins’ of Human Cells”, 2026-05-05